Polymerase als Lückenfüller
Zielgerichtete Mutagenese, Teil 2
Gabriele Grabowski
Die Oligos sind da, die Mutagenese kann endlich losgehen. Gabriele Grabowski beschreibt im zweiten Teil der Mutagenese-Serie wie man mit thermolabilen DNA-Polymerasen zielgerichtet Mutationen in DNA-Stränge einführt.
Alle Primer-Extension-Mutagenesen funktionieren nach einem mehr oder weniger einheitlichen Schema. Neben einer thermolabilen DNA-Polymerase und dNTPs braucht man dazu im einfachsten Fall ein passendes Oligonukleotid (siehe dazu "Zielgerichtete Mutagenese, Teil 1" in
Laborjournal 12/2005, Seite 60-61) sowie eine Matrizen-DNA, die sowohl einzel- als auch doppelsträngig sein kann. Eine geeignete Matrizen-DNA, in der Regel ein rekombinantes Plasmid, das als Mutagenese-Vektor dient, kann man auf verschiedene Arten herstellen:
- Der gewählte Vektor enthält neben den "normalen" Plasmidfunktionen auch den Replikationsursprung filamentöser Phagen (üblicherweise den f1-Ori), es handelt sich also genau genommen um einen Phagemid. In diesem Fall erhält man durch Coinfektion vektorhaltiger Zellen mit Helferphagen einen der beiden DNA-Stränge. (Bei der Oligobestellung auf die Orientierung des Phagen-Oris achten!)
- Das Plasmid wird mithilfe eines Restriktionsenzyms vorlinearisiert. Die Schnittstelle sollte dabei möglichst weit von der zu mutierenden Position entfernt liegen.
- Einige Mutagenese-Verfahren verwenden auch einfach das native, doppelsträngige Molekül.
Die einzelnen Arbeitsschritte
Ist die Matrize fertig, kann es weiter gehen: Matrizen-DNA und Oligo(s) zusammen geben, doppelsträngige Strukturen bei 95 °C bis 100 °C denaturieren und den Ansatz langsam abkühlen lassen (am Besten einfach den Thermoblock ausschalten). Die Oligos müssten jetzt an die Matrize gebunden sein. Anschließend DNA-Polymerase und dNTP's dazu pipettieren. Die Polymerase verlängert die Oligos, die als Primer fungieren, komplementär zur Sequenz des Matrizenstranges ("Fill in"-Reaktion). Die verbleibenden DNA-Enden verknüpft schließlich eine Ligase. Wer will, kann Polymerase und Ligase auch gleichzeitig in den Ansatz geben. Heraus kommen sollte bei beiden Ansätzen ein Heteroduplex, der sich aus einem Strang mit der ursprünglichen Sequenz ("Wildtyp") und einem mit der mutierten Sequenz ("Mutante") zusammensetzt. Die Heteroduplex-DNA schleust man anschließend in Bakterienzellen ein, die das mutierte Plasmid vervielfältigen.
Graue Theorie
Theoretisch müsste jedes zweite vermehrte Plasmid die Mutation tragen - vorausgesetzt, die transformierten Bakterien halten sich an die Lehre von der semikonservativen Replikation der DNA. Das tun aber nur die wenigsten. Wer schon einmal eine Mutagenese versucht hat, weiß, dass
E. colis von dieser Theorie offensichtlich noch nie etwas gehört haben. Darüber hinaus beheben diverse DNA-Reparaturmechanismen in den Zellen Basenfehlpaarungen. Vervielfältigen Sie das mutierte Plasmid deshalb in Bakterienzellen, die kein Mismatch-Reparatur-System enthalten (mutS-Stämme).
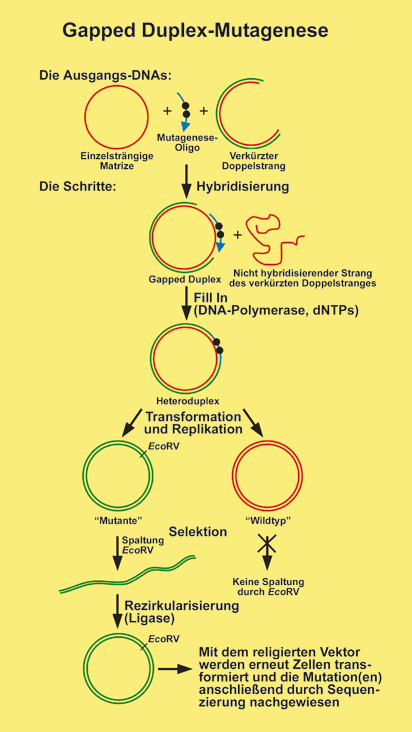
Die Ausbeuten sind oft bescheiden, aber keine Panik, es gibt zahlreiche Tricks, wie man sie erhöhen kann. Wichtig ist, dass die DNA-Polymerase keine allzu langen einzelsträngigen Abschnitte auffüllen muss und wenig zu tun hat. Zusätzliche Mismatches durch unerwünschte Replikationsfehler beschränken sich dadurch auf ein Minimum. Nach diesem Prinzip funktioniert zum Beispiel die Gapped Duplex-Mutagenese (GDM). Bei der GDM enthält der Mutations-Ansatz neben dem Oligo und einem einzelsträngigen Matrizenstrang zusätzlich das doppelsträngige Ursprungsplasmid dem der zu mutierende DNA-Abschnitt fehlt (diesen Abschnitt schneidet man zuvor mit einem Restriktionsenzym heraus). So erreicht man, dass sowohl das Oligo als auch einer der beiden gekürzten Stränge auf der einzelsträngigen Matrize hybridisieren (siehe Abbildung). Die DNA-Polymerase muss dadurch nur verhältnismäßig kurze, einzelsträngige Lücken (Gaps) schließen. Bei den weiteren Schritten sind Transformations- und Plasmidpräparationskünste gefordert. Zunächst schleust man die mutierten Plasmide in Bakterien ein, vermehrt die Bakterien und isoliert anschließend deren Plasmid-DNA. Mutierte Plasmide lassen sich mit dem Restrik-tionsenzym
EcoRV linearisieren und durch Gelelektrophorese von dem nichtgeschnittenen Wildtyp-Plasmid trennen. Jetzt sind Sie Ihrem Ziel schon ziemlich nah. Sie müssen nur noch die Enden der mutierten, linearisierten DNA verknüpfen, das resultierende Plasmid wieder in Zellen einschleusen (dieses mal aber keine mutS-Stämme verwenden), vervielfältigen und erneut isolieren. Vergessen Sie nicht das mutierte Plasmid anschließend zu sequenzieren, um sicher zu sein, dass die gewünschte Mutation auch tatsächlich vorhanden ist.
Oligos clever einsetzen
Bei der oben beschriebenen Gapped Duplex-Mutagenese führt das Oligo gleichzeitig die Mutationen und die Selektionsmöglichkeit durch Restrik-tionsspaltung ein. Falls das nicht möglich ist oder der Phagen-Ori fehlt, bietet sich die sogenannte Transformer Site-Directed Mutagenesis (erhältlich als Kit) als Alternative an. Bei dieser Variante sind zwei getrennte Oligos für Mutagenese und Selektion verantwortlich. Das Selektions-Oligo sollte immer eine bereits vorhandene singuläre Schnittstelle zerstören. Dann kann man sich eine Linearisierung des Ausgangsplasmides oder eine Einzelstrangpräparation komplett ersparen. Im Denaturierungsschritt erfolgt zwar keine vollständige Strangtrennung und während der Hybridisierung renaturieren zahlreiche Wildtyp-Plasmide. Das ist aber kein Problem. Denn verdaut man im Anschluss an die Auffüll-/Ligationsreaktion die DNA mit dem Enzym, dessen Schnittstelle durch das Selektions-Oligo zerstört wurde, dann wird aus der zirculären, renaturierten Ausgangs-DNA wieder lineare DNA. Transformiert man Bakterien mit diesem Ansatz, so verkleinert sich der Wildtyp-Hintergrund, weil Bakterienzellen lineare DNA nur sehr schlecht aufnehmen.
DpnI darf beim Zerschnippeln von Plasmiden mit Restriktionsenzymen natürlich nicht fehlen. Das Enzym erkennt die Sequenz GATC, schneidet im Gegensatz zu anderen Typ II-Restriktionsendonukleasen die DNA aber nur dann, wenn bei dieser Adenin methyliert ist. Nicht methylierte Sequenzen spaltet
DpnI gar nicht, hemimethylierte "nickt" es ausschließlich im methylierten Strang. Die außergewöhnliche
DpnI-Aktivität kann man sich für die Selektion zunutze machen, sofern das Ausgangsplasmid aus einem Stamm isoliert wurde, der die dam-Methylase synthetisiert. Diese methyliert Adenin im Sequenzmotiv GATC. Nach dem Polymerase/Ligase-Schritt ist die gebildete Heteroduplex lediglich im Matrizenstrang methyliert.
DpnI "nickt" deshalb nur diesen Strang mehrfach im Abstand von etwa 200 bis 300 Nukleotiden. Den "genickten" Wild-Typ-Strang replizieren die Zellen jedoch deutlich langsamer als den intakten, mutierten Strang.
Die Replikation des Matrizenstranges lässt sich auch auf andere Weise verhindern, etwa mit der Kunkel-Methode. Bei dieser isoliert man die einzelsträngige DNA aus einem Bakterienstamm dem sowohl die dUTPase als auch die Uracil-N-Glycosylase fehlt (dut-, ung-). Fehlt die dUTPase, so erhöht sich der intrazelluläre dUTP-Spiegel mit der Folge, dass dut--Zellen verstärkt dUTP anstelle von dTTP in die DNA einbauen. Da den Zellen auch die Uracil-N-Glycosylase (ung) fehlt, können sie diesen Fehler nicht korrigieren.
Genickte DNA
Die so gewonnene uracilsubstituierte Matrizen-DNA setzt man dann wie oben beschrieben für die Mutagenese ein und transferiert das resultierende Plasmid in Bakterien, die dut
+ und ung
+ sind. Diese bauen den Matrizenstrang der Heteroduplex bevorzugt ab und hemmen dadurch dessen Replikation. Den neu synthetisierten, mutierten Strang, der keine Uridylate enthält, replizieren sie dagegen ganz normal. Die Selektion findet bei der Kunkel-Methode also direkt in den Zellen statt und benötigt keine zusätzliche enzymatische Vorbehandlung der DNA
in vitro.
Eine Variante der Kunkel-Methode ist die Closing-Oligo-Mutagenese. Diese verwendet das doppelsträngige, uracilsubstituierte Plasmid als Matrize, ein Phagen-Ori ist nicht nötig. Zusätzlich linearisiert man das Plasmid mithilfe eines Restriktionsenzyms und setzt neben dem Mutagenese-Oligo ein weiteres so genanntes Closing-Oligo ein. Dieses hybridisiert auf den freien Enden eines der beiden Stränge des linearisierten Plasmides und erlaubt so dessen gezielte Rezirkularisierung.
Es geht aber auch ganz ohne DNA-Verdau. Dazu braucht man lediglich ein Plasmid das zwei Antibiotika-Resistenzgene enthält. Der Trick besteht darin, dass eines der Resistenzgene für ein intaktes und das andere (aufgrund einer Punktmutation) für ein nichtfunktionierendes Protein kodiert (erhältlich als "Altered Sites II"-System). Gängig ist zum Beispiel die Kombination aus funktionellem Tetracyclin-Resistenzgen (TetR) und inaktiviertem Ampicillin-Resistenzgen (AmpS). Für die Mutagenese verwendet man dieses Plasmid gemeinsam mit drei Oligos. Dabei fungiert nur eines der Oligos als Mutagenese-Oligo, die beiden anderen hybridisieren in jeweils einem der Resistenzgene und führen dort eine neue Punktmutation ein. Diese inaktiviert das Tetracyclin-Resistenzgen, aus TetR wird TetS und reaktiviert das Ampicillin-Resistenz-Gen, aus AmpS wird AmpR. Bei der Altered Sites Mutagenese wird also zwischen den vorhandenen Resistenzen "umgeschaltet". Praktischer Nebeneffekt: Wenn man jeweils zwischen den Resistenzen hin und her wechselt, lassen sich mehrere Mutagenesen in Folge durchführen.
So viel zu den Möglichkeiten von Primer-Extension-Mutagenesen mit thermolabilen DNA-Polymerasen. Im dritten Teil dieser kleinen Serie sind dann PCR basierende Mutagenese-Methoden an der Reihe.
Letzte Änderungen: 16.08.2006